脊肌萎缩症(SMA)
作者:秋凉 | 发布日期:脊肌萎缩症简明指南
脊肌萎缩症是一种进行性下运动神经元损害疾病,主要影响脊髓前角细胞。患者的典型表现是进行性发展的肌肉萎缩,通常首先累及四肢,然后逐渐发展到躯干肌,晚期导致吞咽和呼吸困难,呼吸衰竭是患者的主要死亡原因。根据发病时间和疾病的严重程度,临床上通常可以将脊肌萎缩症粗略地分为四种类型。随着致病基因的发现和功能研究,目前更倾向于认为不同亚型脊肌萎缩症之间并不存在明确的界限,而是疾病有所差异的表现。
脊肌萎缩症是相对常见的遗传病,人群发病率在1/10000左右,人群携带率为1/40-1/60。编码运动神经元存活基因的SMN1基因突变是已知导致脊肌萎缩症的主要原因。脊肌萎缩症目前尚无法治愈。2017年,FDA已经批准基因治疗药物Spinraza上市用于治疗SMA患者。该药物在前期的临床试验中能够非常明显改善I型SMA患者的症状。该药物目前尚未在国内上市。运动康复和对症治疗是延缓病情发展、提高患者生存质量的主要手段。
脊肌萎缩症是一种典型的常染色体隐形遗传病。如果夫妇双方都为携带者,则有25%的几率生育脊肌萎缩症患者,50% 几率生育携带者,25%的几率生育完全正常的孩子。大多数脊肌萎缩症患者的突变SMN1基因遗传自父母,但是有约2%的脊肌萎缩症来自于新发的SMN1基因突变。
SMN1基因突变检测是诊断脊肌萎缩症的最佳方法。95%的脊肌萎缩症患者由SMN1基因纯合缺失导致。通过PCR/RFLP、定量PCR和MLPA等方法对SMN1基因7号外显子进行检测都能够准确检测出SMN1基因纯合缺失从而做出诊断。另一些患者的SMN1基因突变形式并非是纯合缺失,而是缺失了一个SMN1基因同时携带了一个点突变的SMN1基因。这些患者通过SMN1拷贝数定量检测表现为杂合缺失,可以通过进一步对外显子测序找到基因上的其他突变。
通过定量手段开展的SMN1携带者检测能够检出人群中95%的脊肌萎缩症携带者。这不但是确定患者父母携带状态的手段,也是人群筛查的有效方法。但是,部分人的两个SMN1基因位于同一条染色体上,这种特殊的携带者和点突变携带者无法通过定量技术检出,因此有5%的携带者是无法通过常规筛查方法发现的。
什么是脊肌萎缩症
脊肌萎缩症(Spinal Muscular Atrophy,简称SMA )是最常见的常染色体隐性遗传病之一。脊肌萎缩症有时候也被称为“进行性脊肌萎缩症”或者“脊髓性肌萎缩”。脊肌萎缩症的人群发病率约为1/10000。脊肌萎缩症的发病原因在于遗传物质缺陷导致运动神经元受到损害并进而影响肌肉的收缩。在神经肌肉遗传病中,脊肌萎缩症属于典型的神经源性损害性疾病。也就是说,虽然脊肌萎缩症患者变现为肢体运动障碍,但是有问题的并不是肌肉,而是控制肌肉的神经。脊肌萎缩症是婴幼儿和儿童期发病的最常见神经源性损害导致运动发育落后的疾病。
虽然人体的所有活动都受大脑指派,然而直接控制肌肉活动的是位于脊髓内的运动神经元。打个比方,如果把大脑的运动中枢比作中央政府,那么脊髓里的运动神经元就好像是地方政府。从脊肌萎缩症的发病原因来看,“中央政府”的运作很正常,大脑照常发出指令指挥肌肉的活动,可是“地方政府”却没有执行上级的指令,导致肌肉无法收到指令。肌肉尽管本身没有什么问题,然而由于没有接收到指令就不能完成收缩和舒张的活动过程,于是脊肌萎缩症患者表现出活动能力的逐步丧失。中国有一句古话,叫“拳不离手,曲不离口”。无论什么东西,不用的时间长了自然就会退步。对于人体也是如此。由于肌肉长期不能正常收缩,久而久之其收缩的功能发生退化,肌肉逐渐失去正常的外形而变得松散、薄弱,形成了所谓的“废用性萎缩”。看上去是肌肉萎缩了,但是问题的根源还是在于神经。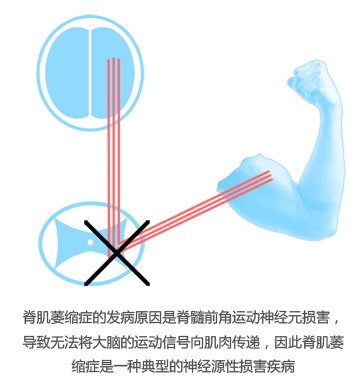
那么,为什么脊肌萎缩症患者的脊髓运动神经元不工作呢?原因在于人体内缺少了一种叫做运动神经元存活蛋白的关键蛋白,这种蛋白对于运动神经元维持正常的功能具有重要作用。脊肌萎缩症患者的运动神经元存活蛋白存在缺陷,使得他们的运动神经元无法正常工作。进一步,这种蛋白缺陷又是从何而来?归根结底,这种蛋白的缺陷正是由编码蛋白的上游基因发生了突变而导致的,这个基因被称为运动神经元存活基因1,简称SMN1。
SMN1基因位于人类的5号染色体上,在正常人体中存在两个相同的SMN1基因,分别位于两条不同的5号染色体上,它们都可以完整地编码“运动神经元存活蛋白”。如果其中一个SMN1基因发生了问题,另一个基因还可以很好地完成自己的任务,起到“补偿”的作用,并不影响肌肉的运动。这种体内带有一个异常SMN1基因的人被称为脊肌萎缩症携带者,他们并不表现出脊肌萎缩症的临床症状,而是表现得和正常人一样。脊肌萎缩症携带者在人群中的频率是1/40-1/60,即大约每50个人中就有一个是脊肌萎缩症携带者。如果人体中的两个SMN1基因都发生了问题,这时候就无法形成完整的运动神经元存活蛋白了。这种具有两个有问题的SMN1基因的人就成为了脊肌萎缩症患者,表现出脊肌萎缩症的临床症状。因此,脊肌萎缩症是一种典型的常染色体隐性遗传病。
脊肌萎缩症的主要症状和临床分型
脊肌萎缩症通常在儿童期发病。脊肌萎缩症患者肌肉张力低下、运动功能受限,因此运动发育显著落后于正常儿童。在别人都会坐的时候还不会抬头、在别人都会走的时候还坐不稳是常见的就诊原因。同时,肌肉运动不良会影响到呼吸、吞咽等重要功能,造成脊肌萎缩症患儿营养不良、呼吸衰竭。误吸、卧床等都是脊肌萎缩症患儿肺部感染的重要诱因,加上患儿本身虚弱的呼吸功能,并且无法有效咳嗽清洁呼吸道,无论小年龄儿童还是年龄更大的患儿,肺部感染和其他呼吸系统并发症都是导致脊肌萎缩症患者死亡的首要原因。对于那些能够存活较长时间的患者而言,脊肌萎缩症带来的运动障碍是必须面对的主要危害,他们必须付出常人不能想象的艰苦努力去努力学习站立、行走、维持受限的运动能力。值得注意的是,脊肌萎缩症并不影响患儿的智力发育,甚至有一些研究还认为脊肌萎缩症患者无论智力、适应性等方面均比人群平均水平更高。因此,对于症状相对较轻的患者而言,脊肌萎缩症并不妨碍他们在一定的领域获得出色的成就。
脊肌萎缩症患者症状的轻重程度存在相当的差异,根据这些症状的严重程度,在临床上可以将脊肌萎缩症分为几种不同的类型。概括来说,发病年龄越早,往往预后越差。对于为什么会产生症状轻重程度的不同,目前认为主要和SMN2基因的表达量有关。如果在基因组中存在较多的SMN2基因,则症状会相对较轻。
1型脊肌萎缩症:有时候也称为婴儿型脊肌萎缩症。通常在6个月以前发病,家长会发现孩子全身松软,双脚活动少,双腿无法抬起,不会蹬被子,哭声较轻,吃奶容易呛到。一些孩子在出生时完全正常,然而在几个月之后逐渐退步,活动逐渐减少。1型脊肌萎缩症是严重的类型,患儿不会独坐,肌肉活动退步很快,逐渐影响到呼吸肌,最后一般都因为肺部感染而死亡,预期寿命通常在2岁以内。
2型脊肌萎缩症:判断2型脊肌萎缩症的主要依据一般是:在7个月至18月之间缓慢发病,患儿能够独坐但是无法独站。其实,2型脊肌萎缩症患儿的症状轻重有很大的不同。与2型脊肌萎缩症患者相比,2型脊肌萎缩症患者的症状总体较轻,存活年限相对较长,但是能存活到青春期的是少数。肌肉活动障碍是相当长一段时间患儿的主要表现,他们的运动能力会在一个较低的平台上停留一段很长的时间而没有明显退步。在年龄增长以后,呼吸、吞咽、脊柱侧弯等问题会逐渐明显。呼吸衰竭依然是患者死亡的主要原因。
3型脊肌萎缩症:3型脊肌萎缩症是轻型的脊肌萎缩症,一般在18月以后发病,也有些患者小时候并没有明显症状,而是在上小学甚至上中学期间因为体育成绩不佳才来就诊。由于这些患者在长大以后以跑不快、上楼困难等原因就诊,常常被误诊为肌营养不良。3型脊肌萎缩症患者可以存活相当长的年限,部分患者的寿命接近正常人。随着年龄的增长,患者的运动能力会缓慢地下降,最终将丧失行走能力,而需要借助轮椅等辅助工具进行活动。
4型脊肌萎缩症:4型脊肌萎缩症是最轻的一种类型,患者一般在20-30岁之间逐渐发病,运动受限很小,发展也比较缓慢。
需要指出的是,脊肌萎缩症的分型是基于临床表现的分型,存在一定的主观性。基因诊断无法帮助分型,同时在疾病的特定阶段可能也无法准确区分临床类型。观察患者的疾病发展过程是分型的唯一依据。病情的严重程度主要取决于肌肉功能丧失的速度,过度纠结于分型并没有实际意义。例如,有些患者可能起病比较早,但是症状比较轻,这时候一定要区分是2型或者3型就显得不那么必要了。
此外,对于脊肌萎缩症的分型随着认识的改变也一直在微调。例如,既往曾经区分一种被称为0型的脊肌萎缩症,主要指患儿在胎儿期就可以受累,出生即表现为全身松软,在数天至数月内死亡的严重类型。目前的分型标准已经不再区分这种类型。就我个人的感觉,在临床上碰到这类严重脊肌萎缩症的可能性也是个案。又如,既往的4型脊肌萎缩症与现在的4型定义也有所区别。我在这里使用的分类标准引自国际协作组在2007年制定的标准。
脊肌萎缩症的临床诊断
脊肌萎缩症的临床诊断主要依靠患者的典型症状、以近端受累为主的运动神经元损害体征以及相应的实验室检查做出。对于脊肌萎缩症患者的诊断主要参照以下表现:
- 典型的临床表现,同时可作为分型参考;
- 下运动神经元损害体征,如肌张力降低、腱反射消失等;
- 肌力下降下肢重于上肢、近端重于远端;
- 一般不伴有上运动神经元损害体征,如病理征等;
- 一般不伴有智力发育落后;
- 肌酶检测在正常范围内或略微偏高。如果CK等肌酶指标显著升高应考虑肌源性疾病;
- 肌电图呈现神经源性损害;
- 肌肉活检呈现典型的神经源性损害改变。
由于脊肌萎缩症是脊髓前角运动神经元受累的疾病,因此掌握脊肌萎缩症的典型表现和下运动神经元受损的体征,对于典型脊肌萎缩症的诊断并不困难。对于患者来说,找到一个对于脊肌萎缩症的诊断富有经验的医生是获得可靠诊断的最便捷途径——简单来说,这篇文章是给医生而不是给患者家属看的。我从来不建议患者和家属去钻研专业教材,医学是专业的,不要否认。疾病的诊断需要医生依靠自己的知识和经验进行综合分析。不要单纯相信某一个检查结果——肌电图可能会不准确,并且经常不准确。
由于基因诊断对脊肌萎缩症的确诊准确可靠,而肌电图比较痛苦,肌肉活检对孩子的伤害更大。因此,在辅助检查手段上,目前一般建议将SMN1基因检测作为脊肌萎缩症诊断的首选方法。
脊肌萎缩症的基因诊断
我们已经说过,脊肌萎缩症是典型的常染色体隐性遗传病,其致病基因是SMN1基因。每个人都有两个SMN1基因,只有两个SMN1基因都发生致病突变才会导致脊肌萎缩症的发生。目前已知SMN1基因的致病突变有两种不同的形式。约95%脊肌萎缩症患者的SMN1基因突变属于“纯合缺失”类型。所谓纯合缺失,是指两个SMN1基因都缺失了,这样身体里就没有SMN1基因,所以会患上脊肌萎缩症。这是SMN1基因突变的常见形式。
另5%左右患者的基因突变不是纯合缺失,而是只缺了一个基因,这种缺失我们叫做“杂合缺失”。按理来讲,杂合缺失的人体内还有一个SMN1基因,他是不会发病的,应该成为无表现的携带者。但是,这些患者体内的那个SMN1不是功能正常的SMN1基因。他们的SMN1基因发生了点突变或者其他形式的突变,使得这个SMN1基因没有功能。这样,尽管他们只缺失了一个SMN1基因,却因为另一个SMN1基因没有功能同样会成为脊肌萎缩症患者。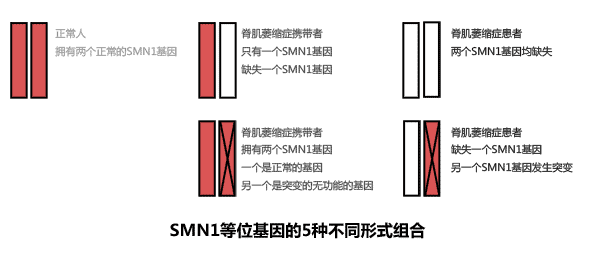
上图为你演示了两种不同的SMN1基因突变。正常人拥有两个正常的SMN1等位基因(红色)。如果缺失一个基因(白色)则为常见的携带者,如果两个等位基因都缺失则成为纯合缺失型患者。如果正常人的一个SMN1基因发生突变(下左图),那么他就是少见的携带者类型。如果缺失一个SMN1基因,又带了一个突变的SMN1基因,就会成为脊肌萎缩症患者。
打个比方,某个保险柜的门上设置了两把锁。如果两把锁都没了,那么保险柜显然也就没有了保险的功能,这就是我们所说的纯合缺失型脊肌萎缩症。如果一把锁没了,而另一把锁换了,保险柜依然没有保险的功能,这就是我们说的突变型脊肌萎缩症。目前,纯合缺失型和突变型脊肌萎缩症在基因层面都可以获得诊断,只是方法的复杂性有所不同。
脊肌萎缩症的基因诊断流程
当医生临床脊肌萎缩症就应该首选SMN1基因纯合缺失检测作为进一步诊断方法。由于SMN1基因检测只需要抽一次血,而且已经成为脊肌萎缩症诊断的标准方法,而肌电图检测患儿较为痛苦同时参考价值相对有限,因此我建议临床疑诊脊肌萎缩症的患者不需要做肌电图检测,而应直接进行基因检测。
SMN1基因纯合缺失检测一般针对SMN1基因的7号外显子进行,以7号外显子是否存在缺失来判断SMN1基因是否缺失,因为7号外显子是SMN1基因的关键区域。纯合缺失检测方法常用的有PCR/RFLP法和荧光定量PCR等。
- PCR/RFLP:PCR/RFLP是经典的SMN1基因7号外显子纯合缺失诊断方法。Dde I与Hinf I是常用的两种内切酶。从方法学上来看,Dde I可能会因为酶切不干净而产生假阴性,Hinf I相对更可靠一些。PCR/RFLP的缺点是无法判断携带者。
- qPCR:荧光定量PCR能够准确分辨患者、携带者和正常人,并且操作简单,是一种拥有更大灵活性的检测方法。
- MLPA:MLPA是一种中通量的检测方法。它的好处是可以通过一个反应检测7号、8号外显子的拷贝数并且检测几个常见点突变位点。MLPA的主要问题是对实验室的质控管理要求较高,它的检测灵活性不及定量PCR。
如果检出SMN1基因7号外显子纯合缺失,可以确诊为脊肌萎缩症,分型需要依赖临床表现和疾病发展进行判断。如果检出患者存在2拷贝SMN1基因(定量PCR法),可基本排除脊肌萎缩症的诊断。
如果患者临床疑诊为脊肌萎缩症,而SMN1基因检测发现患者为杂合缺失,这时候首先建议对患者再次进行临床评估。如果临床症状符合脊肌萎缩症,肌电图等检查也支持诊断,则需要进行SMN1基因外显子测序。由于基因序列的特殊性,SMN1基因测序存在一定的技术难度。常用的方法有以下几种:
- DNA水平普通测序:因为无法排除SMN2基因的干扰,因此DNA水平的外显子测序只能作为筛查方法进行使用。对于DNA水平测序发现突变的患者要做进一步检测确认。
- 长程PCR测序:通过特殊的引物设计将一整段SMN1基因序列扩增出来以去除SMN2基因的影响。长程PCR的问题是扩增片段长,实验不好设计。
- RNA水平测序:通过获得患者RNA进行RT-PCR然后挑单克隆测序是最准确的点突变检测方法。它的缺点是需要用新鲜血液即使抽提RNA,实验比较耗费时间,同时成本较高。
不同的实验室会根据自身实际条件选择适合自己的方法。对于患者和家属来说,你只需要记住这一流程:医生临床诊断脊肌萎缩症,进行SMN1基因纯合缺失检测。如为纯合缺失,则确诊;如为杂合缺失,则再次由医生进行临床评估,并进行SMN1基因点突变检测。并非每一名脊肌萎缩症患者都可以找到基因突变,但是通过这两个步骤,SMN1基因突变的检出率在98%以上。
7号外显子和8号外显子检测有什么区别?

对于SMN1基因缺失检测,有些地方会同时做7号外显子和8号外显子的检测,而有些地方则只做7号外显子的检测。经常有人问我这两者之间有什么区别。上图为你简单展示了SMN1基因的结构。与所有基因相同,SMN1基因也由不同的外显子构成。在SMN1基因中,关键的是7号外显子。如果SMN1基因的7号外显子缺失,即使其他外显子都保存完整,这个SMN1基因依然是没有功能的。而8号外显子并不重要。即使8号外显子缺失了,只要7号外显子完整,这个SMN1基因依然能够正常表达。所以,就SMN1基因的缺失来说,7号外显子是决定性的,在实际检测中只检测7号外显子就可以了。
大多数SMN1基因缺失的人会同时缺失7号和8号外显子,但是也有部分患者只缺失其中一个。需要注意的是,8号外显子纯合缺失不是脊肌萎缩症的诊断依据。就实验来说,同时检测8号外显子有时候有助于验证对7号外显子的判断结果,我认为这对于MLPA检测的意义相对比较重要。然而在判断基因致病性的时候,我们只应该关注7号外显子,而不需要去考虑8号外显子。
脊肌萎缩症的治疗原则
目前,尚没有可以治愈脊肌萎缩症的有效方法。但是医学和技术的进步使得脊肌萎缩症患者的生活质量获得了很大提高。更好的辅助呼吸和喂养技术使得部分原来无法救治的脊肌萎缩症患者的生命得到了延续,而运动医学的发展帮助了大批脊肌萎缩症患者最大可能地保存了一定的运动能力。随着科学技术的不断发展,脊肌萎缩症的治疗方法正在很多实验室进行研究。基因治疗和干细胞移植也许在不久的将来可以为疾病带来治愈的希望。
对于大多数家长来说,不切实际地等待基因和干细胞治疗可能会陷入一些骗局。对于正规教学医院提供的临床试验机会是可以尝试的,然而对于所有收费的研究性治疗则是不推荐的。 期待奇迹的发生是一回事情,然而脚踏实地地面对当前的生活是在期待奇迹的过程中更好的态度。因为,毕竟我们可以做一些事情,让脊肌萎缩症的孩子在疾病无法治愈的情况下,获得更好的生活。
不同类型的脊肌萎缩症患者需要不同的处理思路
如我之前说过的,不同类型的脊肌萎缩症患者在症状的严重程度和预期寿命上存在显著的不同,因此对于不同类型的患者其治疗的侧重点并不一样。1型脊肌萎缩症患者症状最重,预期寿命一般在2岁以内。对于这部分患儿,家长的关注重点是患儿的护理,包括呼吸道的管理以及喂养的管理。运动康复对1型脊肌萎缩症患儿的作用是非常有限的。
2型脊肌萎缩症患儿可以存活较长时间,同时症状的严重程度相差较大,一些孩子可以上小学甚至上中学。对于2型脊肌萎缩症的孩子,运动康复是非常需要的,这可以提高患儿的生活质量,在一定程度上延缓疾病的进展。对于这部分孩子,心理关怀也非常重要。
3型和4型脊肌萎缩症的患者症状较轻,人生的路很长,在运动锻炼、心理辅导及各种支持手段以外,他们所需要学习的东西和常人一般,他们也有能力在自己的领域中获得杰出的成就。对于这些患者,康复的介入非常重要。康复介入越早、越有效,疾病发展得就会越慢。
提高脊肌萎缩症患者生活质量的原则
1、呼吸管理。呼吸道并发症是引起脊肌萎缩症患儿死亡的首要原因,因此一定要注意加强呼吸管理,通过合适的物理和化学的手段保持呼吸道的清洁和通畅。患儿如有呼吸道感染的症状,应及早到医院求医,以免病情加重延误治疗。健康的呼吸功能是脊肌萎缩症患者生存和提高生命质量的关键之一。必要时应寻求呼吸科医生的帮助。通过无创呼吸机辅助通气是目前提高脊肌萎缩症生存率的一个可行方法,这种设备目前在国内也有渠道可以买到。
2、喂养和饮食。对于有吞咽困难的脊肌萎缩症儿童,鼻饲等方法是获得必要营养支持的好方法。避免因吞咽问题而引起的异物吸入,这是引起严重肺炎的原因。而对于症状较为稳定的患者,保证充足的营养、足够的热卡摄入是食谱的关键。高蛋白的瘦肉等食品是保证能量供应的较好选择。
3、运动。适当的运动能够提高患者的生活质量。脊肌萎缩症患者的运动技能通常都经历一个上升、下降、然后在一个水平较长时期维持而没有快速退步的过程。在上升期,通过各种训练可以让患者习得更多的运动技能;而在维持期,在康复科医生或者专科医生的指导下进行全关节范围的主动和被动运动,能够防止脊肌萎缩症患者关节僵硬和尽可能保持肌肉的功能。专门的针对呼吸、吞咽肌肉群的训练对于部分严重病人是必需的。游泳是一个很好的方法,水的浮力可以部分抵消重力,使得患者能够最大限度地完成主动运动。游泳必须要防止溺水以外的发生。良好的运动训练和康复可以最大限度地保留脊肌萎缩症患者的运动能力,避免肌肉的快速萎缩和关节变形,尤其对于能够存活较长时间的较轻症2型和3型脊肌萎缩症患者,将有很大的意义。
4、必要的手术介入。很多脊肌萎缩症患者随着年龄增长脊柱侧弯会日益明显。严重的脊柱侧弯会进一步限制患者的运动能力,并可能影响患者的呼吸。在必要的情况下,可以通过外科手术进行脊柱矫形治疗,以提高患儿的生存质量。
5、心理干预。患儿最终会意识到自己的身体健康存在严重的问题,如何让他们能够更好地面对未来的人生,尤其是对3型脊肌萎缩症患者,如何学会带着疾病继续生活,将是一件非常重要的事情。在这方面,可以寻求心理医生的帮助和指导。
基因治疗
2017年,美国FDA已经批准用于治疗脊肌萎缩症的基因治疗药物Spinraza上市。Spinraza也被称为Nusinersen,这是一种影响SMN基因剪切的反义核苷酸药物。在早先开展的2期临床试验中,研究者报告了出色的疗效。在20名入组的1型脊肌萎缩症患者中,运动能力的改善是显著的。目前,该药物已经在更多类型的SMA患者中开展了研究。远期疗效尚需确认。但是,Spinraza的上市无疑为SMA的治疗开辟了一条新的道路。遗憾的是,目前在国内尚无法使用该药物。
脊肌萎缩症的预防
脊肌萎缩症是一种可以预防的疾病。由于脊肌萎缩症的病因非常明确,因此理论上只需在怀孕期间通过羊水等样本进行SMN1基因纯合缺失检测,即可判断胎儿是否为脊肌萎缩症的患者。目前,这种方法主要针对曾经生育过脊肌萎缩症患儿的父母,起到预防疾病再发的作用。
对于生育过脊肌萎缩症孩子的父母,再生育患儿的风险是1/4。如果本人是脊肌萎缩症携带者,则亲兄弟携带SMN1突变基因的概率大概是1/2。因此,对于已经生育过SMA患者的家长,在再生育时应做好预防工作;对于家族中发现过SMA患者的情况,也应该在怀孕前进行一次携带者筛查,以了解自己的生育风险。
预防脊肌萎缩症再发
对于曾经生育过脊肌萎缩症患儿的父母,在再次怀孕以后,只需在怀孕期间通过绒毛或者羊水进行SMN1基因纯合缺失检测,即可完成产前诊断。这是一个相当简单的过程,但是依然有两点必须注意。
第一,产前诊断必须遵循一般的先证者-携带者-产前诊断的三部曲流程。家长要做的第一件事情是带孩子到医院确诊疾病,并且找到基因突变。然后,夫妇双方需要接受相应的携带者检测以明确携带者状态。最后,在怀孕之后接受羊水SMN1基因检测。很多孕妇甚至医生以为产前诊断就是抽羊水而忽略了之前的两步过程,这是非常危险的。我一直建议当你准备接受产前诊断,一定要在备孕的时候找到明确能够提供脊肌萎缩症产前诊断的单位,询问清除步骤。这里面有很多教训,请一定要记住。
第二件必须注意的事情是,脊肌萎缩症的产前诊断必须针对SMN1基因,做染色体是没用的。很多人把“羊水染色体检查”和“羊水检查”混为一谈,也曾经有人做了一个羊水染色体检查就以为做过了基因检查结果再次生下了患病的孩子。简单说来,羊水只是一个样本的来源,用羊水可以做很多检测,比如染色体,比如各种基因,究竟做什么取决于每个人的实际情况。对于脊肌萎缩症的预防,必须要检测SMN1基因,单纯做染色体检查是无效的。
家族中有脊肌萎缩症患者的情况
脊肌萎缩症是常染色体隐性遗传病,只有两个携带者生育孩子才可能会导致患儿的出生。因此,携带者可能在家族中长期存在而没有家族史。如果你家族中有脊肌萎缩症的患者(例如你兄妹的孩子是脊肌萎缩症,或者你的兄妹中有脊肌萎缩症的患者),那么你本人携带致病基因的概率就较高。假若你的另一半不幸也是携带者的话,那么就有生育脊肌萎缩症孩子的风险。因此,对于家族中曾经诊断过脊肌萎缩症患者的情况,建议本人去做一次脊肌萎缩症携带者检测(只需抽一次血进行SMN1拷贝数检测即可,非常简单)。如果不是脊肌萎缩症携带者,大可不必担心;如果你是脊肌萎缩症携带者,那让你爱人也去做一下。只要你的爱人不是携带者,也不会有什么问题。即使你们两人都携带,只要在怀孕中期通过羊水进行SMN1基因检测,依然可以及早诊断脊肌萎缩症,避免异常患儿的出生。
脊肌萎缩症携带者筛查
夫妇双方均为脊肌萎缩症携带者是生育脊肌萎缩症患儿的前提,那么这种概率有多少呢?在中国人群中,脊肌萎缩症携带者的概率大约是1/40,也就是说每40个人中就有一个是脊肌萎缩症携带者。根据随机的原则,夫妇双方都为脊肌萎缩症携带者的概率是1/1600,每1600对夫妇中就有1对具有生育脊肌萎缩症患儿的风险。进一步,如果某人是脊肌萎缩症携带者,那么他和他的爱人都是脊肌萎缩症携带者的概率就是1/40,这是一个不低的数字。由于脊肌萎缩症的人群携带率较高,部分国家和地区都将脊肌萎缩症列入常规孕前筛查项目。
我认为,脊肌萎缩症携带者筛查是普通夫妇避免脊肌萎缩症患儿出生的一个有效手段,筛查对脊肌萎缩症携带者的检出率达到95%(定量PCR法与MLPA法)。对于有意识的夫妇,可以在备孕时或者孕早期对夫妇中的一人进行脊肌萎缩症携带者检测。如为携带者,则对另一人也进行携带者检测。通过这样的方法,能够避免绝大多数脊肌萎缩症患儿的出生,是一种有效的针对人群的脊肌萎缩症预防措施。
参考阅读
这是我早先翻译的脊肌萎缩症患者Jerry Ferra写给其他SMA患者的一封信,用自己的生活状态来鼓励那些患者的孩子以及他们的家长。宣教手册是我在2010年的时候制作的,虽然时间过去了很久,但是里面大部分内容依然是有参考价值的(除了联系方式以外)。
以下是一些关于脊肌萎缩症的参考资料,主要提供了一些关于疾病与SMN1基因的常用专业文献与工具,仅供专业人员参考。
- OMIM #253300:Spinal Muscular Atrophy, Type 1
- OMIM #253550:Spinal Muscular Atrophy, Type 2
- OMIM #253400:Spinal Muscular Atrophy, Type 3
- OMIM #271150:Spinal Muscular Atrophy, Type 4
- OMIM *600354:Survival of Motor Neuron 1, SMN1
- CCDS 34181: SMN1 Gene, Homo Sapiens
- SMN1 (ENSG00000172062) variation table from 1000 genomes
- Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study